


Contributions of visible and invisible pores to reactive transport in dolomite
Affiliations | Corresponding Author | Cite as | Funding informationPublished by the European Association of Geochemistry
under Creative Commons License CC BY-NC-ND 4.0
Keywords: reactive transport, neutron scattering, CO2 storage, pore scale, carbonates

- Share this article





Article views:377Cumulative count of HTML views and PDF downloads.
- Download Citation
- Rights & Permissions

top
Abstract

Figures
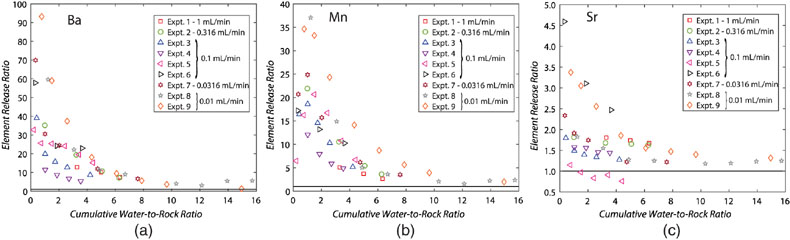 Figure 1 Ca normalised release ratios of (a) Ba, (b) Mn, and (c) Sr during flow-through experiments at flow rates ranging from 0.01 to 1 mL/min, plotted as a function of the cumulative water-to-rock ratio. At the conclusion of the slowest flow rate experiments (0.01 mL/min), ∼60 % of the Ba, ∼30 % of the Mn, and ∼5 % of the Sr had been removed from the cores, while only ∼3 % of the Ca had been removed. | 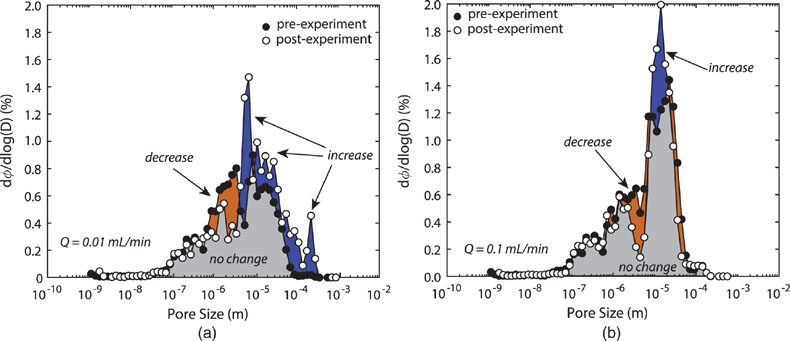 Figure 2 XRCT-USANS-SANS analysis of changes in pore size distribution, represented here as the % of the total sample volume (dφ) that can be attributed to a specific size range of pore sizes (dlog(D)) as calculated by the polydisperse spherical pore (PSDP) model. Integration under the curves yields the entire porosity of the sample. Analysis of pre-experiment (filled circles) and post-experiment (open circles) samples run at flow rates of (a) 0.01 mL/min and (b) 0.1 mL/min permits observation of the decreases and increases of the distribution of pores over specific size ranges. | 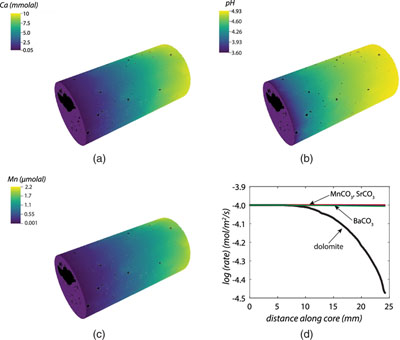 Figure 3 Results of micro-continuum reactive transport simulations on a post-experiment core from a 0.01 mL/min injection experiment. (a) Ca concentration, (b) pH, (c) Mn concentration, and (d) reaction rate in the centre-most element along the length of the core. In (a), (b), and (c), pore space is plotted in black. | 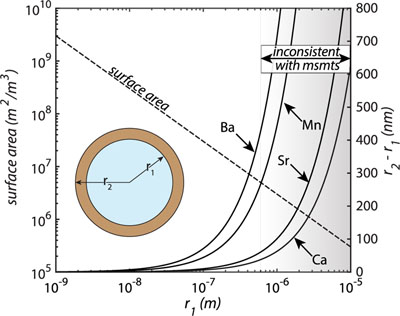 Figure 4 Visualisation of the radius of interaction (r2) required to produce experimentally measured element recoveries for pore radii (r1) ranging from 1 nm to 10 μm and the corresponding specific surface area of these pores. Calculations assume that the rock’s initial porosity is comprised of a uniform distribution of pores of radius = r1. The difference between the trace element (Ba, Mn, Sr) and Ca lines represents the radius from which these elements would have to have been removed without removing Ca to achieve observed recoveries. Figure 2 and accompanying analysis suggests these processes dominantly occurred in pores <600 nm in size, where volumetric specific surface area is orders of magnitude higher and r1–r2 is 10s–100s of times lower than in pores >600 nm in size. msmts, measurements. |
| Figure 1 | Figure 2 | Figure 3 | Figure 4 |
top
Introduction
Carbonate rocks, comprised of calcite, dolomite, and/or aragonite, have been abundant both spatially and temporally throughout Earth’s history. Carbonate reservoirs hold more than 60 % of the world’s remaining oil and 40 % of its natural gas, and have thus historically occupied a significant proportion of the global energy landscape (Tucker and Wright, 2009
Tucker, M.E., Wright, V.P. (2009) Carbonate sedimentology. John Wiley & Sons.
). Looking forward, these same hydrocarbon-trapping characteristics have also made them important targets for geologic carbon dioxide (CO2) storage as the global energy economy transitions away from fossil fuels (Crawshaw and Boek, 2013Crawshaw, J.P., Boek, E.S. (2013) Multi-scale imaging and simulation of structure, flow and reactive transport for CO2 storage and EOR in carbonate reservoirs. Reviews in Mineralogy & Geochemistry 77, 431–458.
).Nonetheless, carbonate rocks are notorious for their often complex pore structures and lack of clear porosity-permeability relationships (Archie, 1952
Archie, G.E. (1952) Classification of carbonate reservoir rocks and petrophysical considerations. AAPG Bulletin 36, 278–298.
; Tucker and Wright, 2009Tucker, M.E., Wright, V.P. (2009) Carbonate sedimentology. John Wiley & Sons.
). To this end, numerous recent studies have examined the hydrogeochemical evolution of carbonates using a combination of imaging and geochemical analyses, often with a specific focus on CO2 injection for geologic CO2 storage (GCS) or Enhanced Oil Recovery (EOR) (Luhmann et al., 2014Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
; Luquot and Gouze, 2009Luquot, L., Gouze, P. (2009) Experimental determination of porosity and permeability changes induced by injection of CO2 into carbonate rocks. Chemical Geology 265, 148–159.
; Smith et al., 2013Smith, M.M., Sholokhova, Y., Hao, Y., Carroll, S.A. (2013) CO2-induced dissolution of low permeability carbonates. Part I: Characterization and experiments. Advances in Water Resources 62, 370–387.
; Wunsch et al., 2013Wunsch, A., Navarre-Sitchler, A.K., Moore, J., Ricko, A., McCray, J.E. (2013) Metal release from dolomites at high partial-pressures of CO2. Applied Geochemistry 38, 33–47.
). This work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013Smith, M.M., Sholokhova, Y., Hao, Y., Carroll, S.A. (2013) CO2-induced dissolution of low permeability carbonates. Part I: Characterization and experiments. Advances in Water Resources 62, 370–387.
; Tutolo et al., 2014Tutolo, B.M., Luhmann, A.J., Kong, X.Z., Saar, M.O., Seyfried, W.E. (2014) Experimental observation of permeability changes in dolomite at CO2 sequestration conditions. Environmental Science and Technology 48, 2445–2452.
), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013Navarre-Sitchler, A.K., Maxwell, R.M., Siirila, E.R., Hammond, G.E., Lichtner, P.C. (2013) Elucidating geochemical response of shallow heterogeneous aquifers to CO2 leakage using high-performance computing: Implications for monitoring of CO2 sequestration. Advances in Water Resources 53, 45–55.
; Wunsch et al., 2013Wunsch, A., Navarre-Sitchler, A.K., Moore, J., Ricko, A., McCray, J.E. (2013) Metal release from dolomites at high partial-pressures of CO2. Applied Geochemistry 38, 33–47.
; Luhmann et al., 2014Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
). Many of these studies have observed non-stoichiometric mobilisation of trace elements during carbonate dissolution, where observed concentrations of trace elements are as much as an order of magnitude higher than would be expected from measured Ca or Mg concentrations. Some evidence demonstrating the exceptional mobility of such trace elements in pure calcite crystals is available in the literature (e.g., Stipp et al., 1992Stipp, S.L., Hochella, M.F., Parks, G.A., Leckie, J.O. (1992) Cd2+ uptake by calcite, solid-state diffusion, and the formation of solid-solution: Interface processes observed with near-surface sensitive techniques (XPS, LEED, and AES). Geochimica et Cosmochimica Acta 56, 1941–1954.
); nevertheless, the pore-scale mechanisms for trace element mobilisation remain poorly constrained.With analytical revolutions over the past several decades, our ability to analyse geochemical processes at smaller and smaller scales has grown, such that processes which were largely theoretically inferred in the final decades of the 20th century are now routinely observable in laboratories around the world. This analytical revolution has led to the understanding that pore-scale processes, i.e. those occurring at the scale of individual pores in a rock, in fact govern the bulk (i.e. continuum-scale) geochemical interactions between rocks and fluids (e.g., Li et al., 2006
Li, L., Peters, C.A., Celia, M.A. (2006) Upscaling geochemical reaction rates using pore-scale network modeling. Advances in Water Resources 29, 1351–1370.
; Molins et al., 2012Molins, S., Trebotich, D., Steefel, C.I., Shen, C. (2012) An investigation of the effect of pore scale flow on average geochemical reaction rates using direct numerical simulation. Water Resources Research 48, 1–11.
; Plümper et al., 2017Plümper, O., Botan, A., Los, C., Liu, Y., Malthe-Sørenssen, A., Jamtveit, B. (2017) Fluid-driven metamorphism of the continental crust governed by nanoscale fluid flow. Nature Geoscience 10, 685–690.
; Ma et al., 2019Ma, J., Querci, L., Hattendorf, B., Saar, M.O., Kong, X.Z. (2019) Toward a Spatiotemporal Understanding of Dolomite Dissolution in Sandstone by CO2-Enriched Brine Circulation. Environmental Science & Technology 53, 12458–12466.
). Indeed, some have argued that the study of pore-scale geochemical processes should be considered a discipline in its own right (Steefel et al., 2015Steefel, C.I., Emmanuel, S., Anovitz, L.M. (Eds.) (2015) Pore-scale geochemical processes. Reviews in Mineralogy & Geochemistry, Volume 80. The Mineralogical Society of America, Virginia, USA.
). This newfound recognition of the importance of pore-scale geochemical processes, combined with our growing knowledge of the size distribution of pores in geologic media (e.g., Anovitz and Cole, 2015Anovitz, L.M., Cole, D.R. (2015) Characterization and analysis of porosity and pore structures. Reviews in Mineralogy and Geochemistry 80, 61–164.
), has led to the question: what is the relative reactivity of a rock’s various pore sizes? In other words, although we now understand the importance of geochemical processes occurring at the scale of individual pores, we do not yet have a quantitative understanding of the relative role of different pore sizes in controlling bulk, continuum-scale geochemical processes. Here, we present new multiscale neutron- and x-ray-based analyses of dolomite rock cores exposed to hydrothermal experiments to address the question of how the various pores sizes present in dolomite rocks impact their bulk hydrogeochemical evolution.top
Methods and Results
Details of hydrothermal flow-through experiments were previously provided by Luhmann et al. (2014)
Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
, but are summarised here to provide context for the presented, new results. Nine experiments were performed on ∼1.3 cm (diameter) × 2.6 cm (length) dolomite cores at 100 °C and 150 bar pore fluid pressure and 200 bar confining pressure to simulate fluid-rock interaction in high pCO2 carbonate reservoirs. Injected solutions contained 1 molal NaCl and 0.65 molal CO2. Experiments lasted from 61 minutes to 9 days, with the duration being a function of the injection rate and the time which it took for permeability to begin to increase exponentially.A wide variety of geochemical techniques suggest that the cores used in our experiments are almost entirely comprised of nearly stoichiometric dolomite, and electron microprobe (EMP) analyses confirm that the dolomite is geochemically homogeneous at the scale of interrogation, i.e. at mapping resolutions of ∼2 μm with a beam diameter of 10 μm (Luhmann et al., 2014
Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
). Spot analyses of Sr and Mn are in good agreement with the bulk geochemical analysis, again confirming that the measured trace elements are embedded within the dolomite structure rather than present as a discrete phase. Assuming that the measured cations are incorporated within the dolomite structure gives the formula: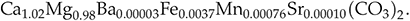
In spite of the homogeneous distribution of trace elements Ba, Mn, and Sr, their concentrations in effluent fluids far exceeded that expected from stochiometric dolomite dissolution in all samples. In the present study, we calculate the Ca normalised release ratio of trace element i from the core according to:
Eq. 1
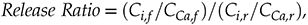
where C indicates concentration, and the subscripts f and r indicate fluid and rock, respectively. Ca was used for these calculations, but virtually identical results would be achieved if Mg was used, since Ca and Mg concentrations were very similar in all samples. Plotting the release ratios against the cumulative water-to-rock ratio (calculated by normalising the cumulative mass of injected water by the mass of the core) demonstrates that ∼10× the amount of most trace cations in the cores, as compared to the major dolomite cations, was recovered during the dissolution experiments, with Ba and Mn being extracted at significantly faster rates than Sr (Fig. 1). The non-stoichiometric dissolution of trace elements suggests that the dissolution process continued even after the solution approached equilibrium with respect to dolomite. However, core-scale porosity changes, as measured by XRCT, agree well with the volumetric removal of dolomite calculated from effluent Ca and Mg concentrations (Luhmann et al., 2014
Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
). XRCT imagery also demonstrates that porosity changes during the experiments occurred largely in channels connecting large pores in the fluid flow path, and, to some degree, augmented pre-existing pores (see Graphical Abstract, caption in Supplementary Information).
Figure 1 Ca normalised release ratios of (a) Ba, (b) Mn, and (c) Sr during flow-through experiments at flow rates ranging from 0.01 to 1 mL/min, plotted as a function of the cumulative water-to-rock ratio. At the conclusion of the slowest flow rate experiments (0.01 mL/min), ∼60 % of the Ba, ∼30 % of the Mn, and ∼5 % of the Sr had been removed from the cores, while only ∼3 % of the Ca had been removed.
The present study was initiated to provide insight into the roles of specific pore sizes in increasing sample porosity/permeability and facilitating trace element recovery. We hypothesised that pores at scales invisible in the XRCT imagery allowed the reactant brines to penetrate into the dolomite and selectively remove the minor, less compatible elements without dissolving significant amounts of the dolomite itself. To test this hypothesis, we performed a series of Small and Ultra-Small Angle Neutron Scattering ((U)SANS) measurements to complement the previously published XRCT imagery. More information about these analyses is given in the Supplementary Information, however, briefly, (U)SANS measurements permit quantitative characterisation of pores with diameters from ∼1 nm to ∼10 μm. These measurements were combined with a re-analysis of the XRCT imagery (8 μm voxel size) to provide a quantitative description of the changes in porosity and pore size distribution over all relevant pore sizes. We chose two end member experiments representative of flow rates of 0.01 and 0.1 mL/min (Expts. 3 and 8, respectively, from Luhmann et al., 2014
Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
) and two “pristine” samples from the same hand sample for the (U)SANS measurements.With the addition of the pore size distribution calculations based on the combined XRCT-USANS-SANS data set, we can delineate which pores were participating in individual physical and chemical processes during the experiments (Fig. 2). Approximately half of the total pore volume in the dolomite cores occurs at pore sizes <8 μm, and is thus invisible at the scale of our XRCT measurements (Table S-1). Pores 600 nm–5 μm grew to >5 μm, while pores <600 nm showed no change, with slower flow rates yielding more, larger porosity. The combined chemical and physical analyses demonstrate that the largest (≥5 μm) pores dominantly contribute to changes in porosity and permeability, consistent with these pores providing access to through-flowing fluids. However, these analyses and calculations of required interaction volumes (Supplementary Information) also strongly suggest that the smallest (≲600 nm) pores actively interact with the larger (>600 nm) pores, in turn suggesting that they greatly contribute to the non-stoichiometric release of trace elements, such that fluids are effectively interacting with almost the entire rock volume.

Figure 2 XRCT-USANS-SANS analysis of changes in pore size distribution, represented here as the % of the total sample volume (dφ) that can be attributed to a specific size range of pore sizes (dlog(D)) as calculated by the polydisperse spherical pore (PSDP) model. Integration under the curves yields the entire porosity of the sample. Analysis of pre-experiment (filled circles) and post-experiment (open circles) samples run at flow rates of (a) 0.01 mL/min and (b) 0.1 mL/min permits observation of the decreases and increases of the distribution of pores over specific size ranges.
top
Reactive Transport Modelling of Steady State Trace Element Fluxes
The new observations were incorporated into a PFLOTRAN (Lichtner et al., 2019
Lichtner, P.C., Hammond, G.E., Lu, C., Karra, S., Bisht, G., Andre, B., Mills, R.T., Kumar, J., Frederick, J.M. (2019) PFLOTRAN Web page. http://www.pflotran.org.
) micro-continuum reactive transport model in order to test our hypothesis regarding trace element mobility during carbonate interactions with CO2-rich fluids. A description of this model and its inputs is included in the Supplementary Information. In the case of dolomite dissolution, the results of the model demonstrate excellent agreement with output fluids sampled by Luhmann et al. (2014)Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
(Fig. 3). Moreover, the results also demonstrate a reduction in the average (i.e. continuum-scale) rate of dolomite dissolution as the solution approaches equilibrium with respect to dolomite, but a near constant average dissolution rate of the trace components because the solution remains far from equilibrium with respect to these phases (Fig. 3d). In the case of the simulated trace components (Mn, Sr, and Ba), the outlet concentrations are ∼10 % of the measured values i.e. 2.2 μmolal versus ∼15–16 μmol/kg in the case of Mn (Fig. 3c); 0.18 μmolal versus 1.5 μmol/kg in the case of Ba, and 0.37 μmolal versus 1.1 μmol/kg in the case of Sr. Because solid state diffusion rates of trace elements in carbonates at low temperatures only permit Angstrom-scale transport distances over our minute-to-hour fluid residence times (Stipp et al., 1992Stipp, S.L., Hochella, M.F., Parks, G.A., Leckie, J.O. (1992) Cd2+ uptake by calcite, solid-state diffusion, and the formation of solid-solution: Interface processes observed with near-surface sensitive techniques (XPS, LEED, and AES). Geochimica et Cosmochimica Acta 56, 1941–1954.
), this observation demands that the reaction rates of these trace components are ∼10× faster than that of dolomite.
Figure 3 Results of micro-continuum reactive transport simulations on a post-experiment core from a 0.01 mL/min injection experiment. (a) Ca concentration, (b) pH, (c) Mn concentration, and (d) reaction rate in the centre-most element along the length of the core. In (a), (b), and (c), pore space is plotted in black.
The enhanced trace element reaction rates can be explained in two ways. Either: 1) the reaction rate of these trace components is significantly faster than that of the bulk dolomite because of the inherent energetic unfavourability of these elements in the dolomite structure (Marini, 2006
Marini, L. (2006) Geological sequestration of carbon dioxide: Thermodynamics, kinetics, and reaction path modeling. Developments in Geochemistry, volume 11. Elsevier, Amsterdam, Oxford.
); or 2) these trace elements are dissolving from nanoscale pores with a reactive surface area ∼10× greater than the dominant flow paths. In reality, each of these factors probably plays a role, but the latter is more strongly supported when considering not only the simulated, steady state reaction rates (to which the former likely also contributes) but also the extent of reaction in the dolomite surrounding these pores required to achieve such high trace element recoveries (Fig. 4). By virtue of their minute size, these pores have a much larger surface area-to-volume ratio and much smaller required interaction radii than their larger counterparts (Fig. 4), which would serve to facilitate both rapid dolomite equilibration and/or recrystallisation as well as rapid trace element extraction.
Figure 4 Visualisation of the radius of interaction (r2) required to produce experimentally measured element recoveries for pore radii (r1) ranging from 1 nm to 10 μm and the corresponding specific surface area of these pores. Calculations assume that the rock’s initial porosity is comprised of a uniform distribution of pores of radius = r1. The difference between the trace element (Ba, Mn, Sr) and Ca lines represents the radius from which these elements would have to have been removed without removing Ca to achieve observed recoveries. Figure 2 and accompanying analysis suggests these processes dominantly occurred in pores <600 nm in size, where volumetric specific surface area is orders of magnitude higher and r1–r2 is 10s–100s of times lower than in pores >600 nm in size. msmts, measurements.
Together, our results can be used to inform a conceptual model for the role of a rock’s various pore sizes in its hydrogeochemical evolution that assigns a specific role to both the “visible” and “invisible” pores. In this conceptual model, fluids in the “visible” pores remain in a dissolution dominated, pore enlarging regime and probably do not contribute much more than the stoichiometrically expected mass of trace elements to the through-going fluids, since doing so would require extreme interaction volumes (Fig. 4). At the same time, fluids in “invisible” pores approach equilibrium with respect to primary dolomite, but remain far from equilibrium with respect to its trace components. The saturation states of the fluids in these pores thus allow the fluid to extract the trace elements without significantly increasing the pore size. This could be achieved through selective extraction of these elements from the dolomite structure due to structural misfit, or from dissolution of primary dolomite and re-precipitation of a purer phase. Although structural misfit is a plausible explanation for more rapid, instantaneous surface reaction rates, consideration of the interaction radii plotted in Fig. 4 (up to 10s to 100s of 15.95 Å; Steinfink and Sans, 1959
Steinfink, H., Sans, F.J. (1959) Refinement of hte crystal structure of dolomite. American Mineralogist 44, 679–682.
) dolomite unit cells for “invisible” (<600 nm) pores) and the lack of change in the proportion of pores <600 nm in size suggests that coupled dolomite dissolution-reprecipitation likely plays an important role. Regardless of the mechanism, the elevated recoveries demonstrate that the “invisible” pores are connected to the larger scale “visible” flow paths in the core, such that the fluids are efficiently leaching trace elements from a dense, nanoporous network, but only altering porosity in select core sections.top
Implications
The results presented here have significant implications for both the development of hydrogeochemical models of sedimentary rocks as well as the use of carbonate rocks for the interpretation of past environmental conditions. We have shown that a very significant fraction of pores within carbonate rocks are present at scales that are not easily probed using conventional imaging techniques. Our combined analyses have shown that the largest pores in our samples contribute to the dissolution-driven porosity increases but that the smallest pores are most likely contributing to trace element release from the rock without significantly changing in size. This latter observation indicates that trace elements can be removed from dolomite rocks without leaving important telltale signs of alteration, such as pore enlargement or mineral etching. Increasingly, the trace element contents of ancient carbonate rocks are being used to interpret past Earth conditions (e.g., Gilleaudeau et al., 2016
Gilleaudeau, G.J., Frei, R., Kaufman, A.J., Kah, L.C., Azmy, K., Bartley, J.K., Chernyavskiy, P., Knoll, A.H. (2016) Oxygenation of the mid-Proterozoic atmosphere: Clues from chromium isotopes in carbonates. Geochemical Perspectives Letters 2, 178–187.
; Liu et al., 2016Liu, X.M., Kah, L.C., Knoll, A.H., Cui, H., Kaufman, A.J., Shahar, A., Hazen, R.M. (2016) Tracing Earth’s O2 evolution using Zn/Fe ratios in marine carbonates. Geochemical Perspectives Letters 2, 24–34.
), yet our results show that the trace element content of these rocks can be very dramatically altered during water-rock interaction over geologic time. Although our experiments were run with CO2 concentrations higher than may be encountered in most carbonate reservoirs, they were also extremely short in duration when compared with diagenetic processes occurring over geologic time.top
Acknowledgements
We thank the three anonymous reviewers for their insightful comments that helped to improve the clarity of the manuscript. Dr. Xin Gu (Pennsylvania State University) provided scripts for processing autocorrelation data and Hanford Deglint (University of Calgary) helped in creating Figure 2. Research support was provided by the National Science and Engineering Research Council of Canada (RGPIN-2018-03800), the Donors of the American Chemical Society Petroleum Research Fund, and the University of Calgary. MOS and X-ZK thank the Werner Siemens-Stiftung (Werner Siemens Foundation) for their support of the Geothermal Energy and Geofluids (GEG.ethz.ch) group at ETH Zurich, Switzerland. NCNR (U)SANS instrumentation was supported in part by the NSF under agreement DMR-0944662, and ISIS beamtime was supported by the Science and Technology Facilities Council of the United Kingdom RB1610074. ISIS SANS data doi: 10.5286/ISIS.E.RB1610074.
Editor: Satish Myneni
top
References
Anovitz, L.M., Cole, D.R. (2015) Characterization and analysis of porosity and pore structures. Reviews in Mineralogy and Geochemistry 80, 61–164.
 Show in context
Show in contextThis newfound recognition of the importance of pore-scale geochemical processes, combined with our growing knowledge of the size distribution of pores in geologic media (e.g., Anovitz and Cole, 2015), has led to the question: what is the relative reactivity of a rock’s various pore sizes?
View in article
Archie, G.E. (1952) Classification of carbonate reservoir rocks and petrophysical considerations. AAPG Bulletin 36, 278–298.
Show in contextNonetheless, carbonate rocks are notorious for their often complex pore structures and lack of clear porosity-permeability relationships (Archie, 1952; Tucker and Wright, 2009).
View in article
Crawshaw, J.P., Boek, E.S. (2013) Multi-scale imaging and simulation of structure, flow and reactive transport for CO2 storage and EOR in carbonate reservoirs. Reviews in Mineralogy & Geochemistry 77, 431–458.
Show in contextLooking forward, these same hydrocarbon-trapping characteristics have also made them important targets for geologic carbon dioxide (CO2) storage as the global energy economy transitions away from fossil fuels (Crawshaw and Boek, 2013).
View in article
Gilleaudeau, G.J., Frei, R., Kaufman, A.J., Kah, L.C., Azmy, K., Bartley, J.K., Chernyavskiy, P., Knoll, A.H. (2016) Oxygenation of the mid-Proterozoic atmosphere: Clues from chromium isotopes in carbonates. Geochemical Perspectives Letters 2, 178–187.
Show in contextIncreasingly, the trace element contents of ancient carbonate rocks are being used to interpret past Earth conditions (e.g., Gilleaudeau et al., 2016; Liu et al., 2016), yet our results show that the trace element content of these rocks can be very dramatically altered during water-rock interaction over geologic time.
View in article
Li, L., Peters, C.A., Celia, M.A. (2006) Upscaling geochemical reaction rates using pore-scale network modeling. Advances in Water Resources 29, 1351–1370.
Show in contextThis analytical revolution has led to the understanding that pore-scale processes, i.e. those occurring at the scale of individual pores in a rock, in fact govern the bulk (i.e. continuum-scale) geochemical interactions between rocks and fluids (e.g., Li et al., 2006; Molins et al., 2012; Plümper et al., 2017; Ma et al., 2019).
View in article
Lichtner, P.C., Hammond, G.E., Lu, C., Karra, S., Bisht, G., Andre, B., Mills, R.T., Kumar, J., Frederick, J.M. (2019) PFLOTRAN Web page. http://www.pflotran.org.
Show in contextThe new observations were incorporated into a PFLOTRAN (Lichtner et al., 2019) micro-continuum reactive transport model in order to test our hypothesis regarding trace element mobility during carbonate interactions with CO2-rich fluids.
View in article
Liu, X.M., Kah, L.C., Knoll, A.H., Cui, H., Kaufman, A.J., Shahar, A., Hazen, R.M. (2016) Tracing Earth’s O2 evolution using Zn/Fe ratios in marine carbonates. Geochemical Perspectives Letters 2, 24–34.
Show in contextIncreasingly, the trace element contents of ancient carbonate rocks are being used to interpret past Earth conditions (e.g., Gilleaudeau et al., 2016; Liu et al., 2016), yet our results show that the trace element content of these rocks can be very dramatically altered during water-rock interaction over geologic time.
View in article
Luhmann, A.J., Kong, X.Z., Tutolo, B.M., Garapati, N., Bagley, B.C., Saar, M.O., Seyfried, W.E. Jr. (2014) Experimental dissolution of dolomite by CO2-charged brine at 100°C and 150 bar: Evolution of porosity, permeability, and reactive surface area. Chemical Geology 380, 145–160.
Show in contextTo this end, numerous recent studies have examined the hydrogeochemical evolution of carbonates using a combination of imaging and geochemical analyses, often with a specific focus on CO2 injection for geologic CO2 storage (GCS) or Enhanced Oil Recovery (EOR) (Luhmann et al., 2014; Luquot and Gouze, 2009; Smith et al., 2013; Wunsch et al., 2013).
View in article
This work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013; Tutolo et al., 2014), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013; Wunsch et al., 2013; Luhmann et al., 2014).
View in article
Details of hydrothermal flow-through experiments were previously provided by Luhmann et al. (2014), but are summarised here to provide context for the presented, new results.
View in article
A wide variety of geochemical techniques suggest that the cores used in our experiments are almost entirely comprised of nearly stoichiometric dolomite, and electron microprobe (EMP) analyses confirm that the dolomite is geochemically homogeneous at the scale of interrogation, i.e. at mapping resolutions of ∼2 μm with a beam diameter of 10 μm (Luhmann et al., 2014).
View in article
However, core-scale porosity changes, as measured by XRCT, agree well with the volumetric removal of dolomite calculated from effluent Ca and Mg concentrations (Luhmann et al., 2014).
View in article
We chose two end member experiments representative of flow rates of 0.01 and 0.1 mL/min (Expts. 3 and 8, respectively, from Luhmann et al., 2014) and two “pristine” samples from the same hand sample for the (U)SANS measurements.
View in article
In the case of dolomite dissolution, the results of the model demonstrate excellent agreement with output fluids sampled by Luhmann et al. (2014) (Fig. 3).
View in article
Luquot, L., Gouze, P. (2009) Experimental determination of porosity and permeability changes induced by injection of CO2 into carbonate rocks. Chemical Geology 265, 148–159.
Show in contextTo this end, numerous recent studies have examined the hydrogeochemical evolution of carbonates using a combination of imaging and geochemical analyses, often with a specific focus on CO2 injection for geologic CO2 storage (GCS) or Enhanced Oil Recovery (EOR) (Luhmann et al., 2014; Luquot and Gouze, 2009; Smith et al., 2013; Wunsch et al., 2013).
View in article
Ma, J., Querci, L., Hattendorf, B., Saar, M.O., Kong, X.Z. (2019) Toward a Spatiotemporal Understanding of Dolomite Dissolution in Sandstone by CO2-Enriched Brine Circulation. Environmental Science & Technology 53, 12458–12466.
Show in contextThis analytical revolution has led to the understanding that pore-scale processes, i.e. those occurring at the scale of individual pores in a rock, in fact govern the bulk (i.e. continuum-scale) geochemical interactions between rocks and fluids (e.g., Li et al., 2006; Molins et al., 2012; Plümper et al., 2017; Ma et al., 2019).
View in article
Marini, L. (2006) Geological sequestration of carbon dioxide: Thermodynamics, kinetics, and reaction path modeling. Developments in Geochemistry, volume 11. Elsevier, Amsterdam, Oxford.
Show in contextEither: 1) the reaction rate of these trace components is significantly faster than that of the bulk dolomite because of the inherent energetic unfavourability of these elements in the dolomite structure (Marini, 2006); or 2) these trace elements are dissolving from nanoscale pores with a reactive surface area ∼10× greater than the dominant flow paths.
View in article
Molins, S., Trebotich, D., Steefel, C.I., Shen, C. (2012) An investigation of the effect of pore scale flow on average geochemical reaction rates using direct numerical simulation. Water Resources Research 48, 1–11.
Show in contextThis analytical revolution has led to the understanding that pore-scale processes, i.e. those occurring at the scale of individual pores in a rock, in fact govern the bulk (i.e. continuum-scale) geochemical interactions between rocks and fluids (e.g., Li et al., 2006; Molins et al., 2012; Plümper et al., 2017; Ma et al., 2019).
View in article
Navarre-Sitchler, A.K., Maxwell, R.M., Siirila, E.R., Hammond, G.E., Lichtner, P.C. (2013) Elucidating geochemical response of shallow heterogeneous aquifers to CO2 leakage using high-performance computing: Implications for monitoring of CO2 sequestration. Advances in Water Resources 53, 45–55.
Show in contextThis work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013; Tutolo et al., 2014), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013; Wunsch et al., 2013; Luhmann et al., 2014).
View in article
Plümper, O., Botan, A., Los, C., Liu, Y., Malthe-Sørenssen, A., Jamtveit, B. (2017) Fluid-driven metamorphism of the continental crust governed by nanoscale fluid flow. Nature Geoscience 10, 685–690.
Show in contextThis analytical revolution has led to the understanding that pore-scale processes, i.e. those occurring at the scale of individual pores in a rock, in fact govern the bulk (i.e. continuum-scale) geochemical interactions between rocks and fluids (e.g., Li et al., 2006; Molins et al., 2012; Plümper et al., 2017; Ma et al., 2019).
View in article
Smith, M.M., Sholokhova, Y., Hao, Y., Carroll, S.A. (2013) CO2-induced dissolution of low permeability carbonates. Part I: Characterization and experiments. Advances in Water Resources 62, 370–387.
Show in contextTo this end, numerous recent studies have examined the hydrogeochemical evolution of carbonates using a combination of imaging and geochemical analyses, often with a specific focus on CO2 injection for geologic CO2 storage (GCS) or Enhanced Oil Recovery (EOR) (Luhmann et al., 2014; Luquot and Gouze, 2009; Smith et al., 2013; Wunsch et al., 2013).
View in article
This work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013; Tutolo et al., 2014), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013; Wunsch et al., 2013; Luhmann et al., 2014).
View in article
Steefel, C.I., Emmanuel, S., Anovitz, L.M. (Eds.) (2015) Pore-scale geochemical processes. Reviews in Mineralogy & Geochemistry, Volume 80. The Mineralogical Society of America, Virginia, USA.
Show in contextIndeed, some have argued that the study of pore-scale geochemical processes should be considered a discipline in its own right (Steefel et al., 2015).
View in article
Steinfink, H., Sans, F.J. (1959) Refinement of hte crystal structure of dolomite. American Mineralogist 44, 679–682.
Show in contextAlthough structural misfit is a plausible explanation for more rapid, instantaneous surface reaction rates, consideration of the interaction radii plotted in Fig. 4 (up to 10s to 100s of 15.95 Å; Steinfink and Sans, 1959) dolomite unit cells for “invisible” (<600 nm) pores) and the lack of change in the proportion of pores <600 nm in size suggests that coupled dolomite dissolution-reprecipitation likely plays an important role.
View in article
Stipp, S.L., Hochella, M.F., Parks, G.A., Leckie, J.O. (1992) Cd2+ uptake by calcite, solid-state diffusion, and the formation of solid-solution: Interface processes observed with near-surface sensitive techniques (XPS, LEED, and AES). Geochimica et Cosmochimica Acta 56, 1941–1954.
Show in contextSome evidence demonstrating the exceptional mobility of such trace elements in pure calcite crystals is available in the literature (e.g., Stipp et al., 1992); nevertheless, the pore-scale mechanisms for trace element mobilisation remain poorly constrained.
View in article
Because solid state diffusion rates of trace elements in carbonates at low temperatures only permit Angstrom-scale transport distances over our minute-to-hour fluid residence times (Stipp et al., 1992), this observation demands that the reaction rates of these trace components are ∼10× faster than that of dolomite.
View in article
Tucker, M.E., Wright, V.P. (2009) Carbonate sedimentology. John Wiley & Sons.
Show in contextCarbonate reservoirs hold more than 60 % of the world’s remaining oil and 40 % of its natural gas, and have thus historically occupied a significant proportion of the global energy landscape (Tucker and Wright, 2009).
View in article
Nonetheless, carbonate rocks are notorious for their often complex pore structures and lack of clear porosity-permeability relationships (Archie, 1952; Tucker and Wright, 2009).
View in article
Tutolo, B.M., Luhmann, A.J., Kong, X.Z., Saar, M.O., Seyfried, W.E. (2014) Experimental observation of permeability changes in dolomite at CO2 sequestration conditions. Environmental Science and Technology 48, 2445–2452.
Show in contextThis work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013; Tutolo et al., 2014), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013; Wunsch et al., 2013; Luhmann et al., 2014).
View in article
Wunsch, A., Navarre-Sitchler, A.K., Moore, J., Ricko, A., McCray, J.E. (2013) Metal release from dolomites at high partial-pressures of CO2. Applied Geochemistry 38, 33–47.
Show in contextTo this end, numerous recent studies have examined the hydrogeochemical evolution of carbonates using a combination of imaging and geochemical analyses, often with a specific focus on CO2 injection for geologic CO2 storage (GCS) or Enhanced Oil Recovery (EOR) (Luhmann et al., 2014; Luquot and Gouze, 2009; Smith et al., 2013; Wunsch et al., 2013).
View in article
This work has generally concentrated on the dissolution of primary carbonate minerals, and coupled changes in porosity and permeability during reaction with dissolved CO2 (Smith et al., 2013; Tutolo et al., 2014), although a number of studies have examined the mobility of trace elements during CO2-driven dissolution of the primary carbonate minerals (Navarre-Sitchler et al., 2013; Wunsch et al., 2013; Luhmann et al., 2014).
View in article
top
Supplementary Information
The Supplementary Information includes:
- Graphical Abstract Caption
- Supplementary Methods
- Figures S-1 and S-2
- Tables S-1 to S-3
- Supplementary Information References
Download the Supplementary Information (PDF)
Figures



